
Artículo de Revisión Front. Neuroanat., 01 de marzo de 2016 Volumen 10 - 2016 | https://doi.org/10.3389/fnana.2016.00013 [1, 2] Este artículo forma parte del Tema de Investigación: Los Grandes Descubrimientos de Cajal y sus Discípulos: Hitos Consolidados para la Neurociencia del Siglo XXI.
Autor: Santiago Ramon y Cajal Agüeras* Departamento de Patología, Hospital Universitario Vall d’Hebron, Universidad Autónoma de Barcelona, Barcelona, España [1, 6]
Resumen
Los últimos 140 años han sido testigos de considerables avances en el conocimiento de los tumores del sistema nervioso central. Sin embargo, los principales tipos de tumores ya habían sido descritos durante los primeros años del siglo XX. Los estudios del Dr. Pío del Río Hortega se cuentan entre los análisis más exhaustivos de los tumores del sistema nervioso basados en la histología y la citología [6]. La obra de Río Hortega se llevó a cabo utilizando métodos de tinción con plata, que requerían un alto nivel de habilidad práctica y, por lo tanto, eran difíciles de estandarizar. Su aptitud técnica y su interés en los tumores del sistema nervioso desempeñaron un papel clave en el establecimiento de su clasificación, que se basaba en el linaje celular y el desarrollo embrionario. El enfoque de Río Hortega fue controvertido cuando lo propuso. Las clasificaciones actuales no solo se basan en el tipo celular y el linaje embrionario, sino también en características clínicas, la localización anatómica y la edad.
Palabras clave: histogénesis, gliomas, Río Hortega, tumores cerebrales, clasificación [7].
Introducción
Es una satisfacción poder resumir la contribución de Pío del Río Hortega al campo de la neuropatología, en particular, a los tumores del sistema nervioso central. Como patólogo, soy plenamente consciente de las clasificaciones de los tumores del sistema nervioso central y puedo ahora proporcionar un contexto para muchas de las aportaciones del Dr. Pío del Río Hortega.
Como se ha señalado en otras secciones de esta monografía, Río Hortega fue un personaje ilustre, tanto a nivel humano como científico. Hombre autodidacta con un extraordinario conocimiento de las técnicas de laboratorio, fue una figura científica destacada en las ciudades donde trabajó. La combinación de pericia, dedicación y una innegable persistencia y capacidad de trabajo, junto con un considerable talento, le llevaron a realizar descubrimientos clave en la historia de la neurociencia, incluyendo la microglía y la oligodendroglía (como se describe en otros capítulos de este Tema de Investigación especial) [6, 8, 9].
Su genio no residía únicamente en la observación, sino en la capacidad de hacer visible lo invisible. Sus técnicas, especialmente el método de tinción con carbonato de plata, le permitieron trabajar sobre los tumores del sistema nervioso central y desarrollar su clasificación, eminentemente práctica [6, 10, 11]. Este dominio metodológico fue la base sobre la que se construyeron sus avances conceptuales, demostrando que en la neuropatología, el acto de ver es inseparable del acto de descubrir.
Contexto Histórico
Las primeras descripciones de tumores cerebrales datan del período de Virchow, quien describió los gliomas como tumores derivados de las células neurogliales. Virchow distinguió entre mixoglioma, gliosarcoma, glioma durum y glioma hemorrhagicum, compuestos por células gliales que a veces contenían fibras. La pionera comparación de Virchow entre las células neoplásicas y el tejido cerebral normal sentó las bases científicas para todas las clasificaciones posteriores de los tumores del sistema nervioso central [6]. Los principales estudios de finales del siglo XIX y principios del XX fueron realizados por Simon (1874), que describió el glioma de células en araña, y por Tooth y Conheim, que postularon que los tumores surgían de restos embrionarios.
Entre 1900 y 1950, las diversas clasificaciones de los tumores del sistema nervioso central generaron décadas de confusión terminológica. Los estudios más notables del período fueron los de Ribbert (1910, 1918), quien especuló sobre la histogénesis y la etiología de los gliomas, particularmente en su artículo sobre el espongioblastoma y el glioma (“Über das Spongioblastoma und das Gliom”) [6]. Algunos autores consideran que este estudio tuvo un efecto negativo en las clasificaciones de gliomas que se elaboraron durante los siguientes 20 años. En su estudio, basado en deducciones teóricas, Ribbert concluyó que las áreas gliales diferenciadas nunca pueden regresar a un grado inferior de diferenciación y que, por lo tanto, los gliomas, glioblastomas y espongioblastomas tendrían que explicarse necesariamente por la presencia de restos embrionarios cuyo crecimiento se había detenido en diversas etapas de diferenciación. Según esta hipótesis, Ribbert creía que los tumores provenían de estadios embrionarios y no de cambios que ocurrían en las células más diferenciadas. No obstante, Ribbert allanó el camino para el estudio citológico de los tumores y para estudios más específicos basados en métodos de impregnación, de los cuales Río Hortega fue un importante defensor. El enfoque histogenético y embriológico adoptado por Ribbert fue modificado por el enfoque celular, defendido principalmente por Río Hortega [6].
Las contribuciones de la escuela francesa (Lhermitte y Dumas, 1916; Cornil, 1924; Roussy y Oberling, 1932) alrededor de la década de 1920 permitieron distinguir entre el astrocitoma fibrilar, cuatro subgrupos de glioma no fibrilar (células redondas, fusiformes, polimórficas y ameboides), el glioblastoma y el espongioblastoma. La clasificación incluía los ependimomas junto con los tumores del plexo coroideo, que estaban separados de los otros gliomas [6]. El enfoque histogenético se mantuvo en los estudios de Globus y Strauss (1925) y en los de Bailey y Cushing (1926), donde se distinguen varios tipos de células histogenéticas en los gliomas. Los autores reconocen la considerable heterogeneidad interna de estos tumores, hasta el punto de que su clasificación ponía un énfasis considerable en el tipo celular predominante.
Los mismos autores realizaron un estudio exhaustivo de los tumores cerebrales basado en características morfológicas y en correlaciones con el pronóstico del paciente tras la cirugía [1]. Su clasificación se desarrolló a partir del concepto de que las células tumorales podían surgir de una célula madre del epitelio medular, que podía diferenciarse en otras células gliales, neurales o coroideas. Estas células podían luego diferenciarse aún más. En teoría, los tumores podían desarrollarse en cada una de estas fases de diferenciación. Este período vio la primera descripción de los oligodendrogliomas y los meduloblastomas cerebelosos. Aunque estos tumores habían sido descritos como sarcomas o neuroblastomas por otros autores, Bailey y Cushing (1925) tienen el mérito de separarlos del neuroblastoma basándose en su apariencia macroscópica, origen, forma de crecimiento y diseminación a lo largo de la médula espinal [6].
La clasificación de Bailey y Cushing de 1926 es similar a la actual. Distinguía entre los tumores del parénquima nervioso central de la siguiente manera:
-
Astrocitoma (grados I–IV), astrocitoma pilocítico, glioblastoma multiforme, oligodendroglioma, ependimoma, papiloma del plexo coroideo, pinealoma, quiste coloide y meduloblastoma.
-
Tumores meníngeos: meningioma, meningioma maligno, sarcoma meníngeo y meningiomatosis.
-
Tumores de los pares craneales: neurinoma.
-
Tumores de la glándula pituitaria: adenoma, adenoma invasivo, carcinoma y craneofaringioma.
-
Tumores vasculares: hemangioblastoma.
-
Tumores embrionarios: quistes dermoides y teratomas.
Esta clasificación tuvo un enorme impacto en la neurociencia y la neurocirugía, aunque fue criticada por varios autores, principalmente Scherer, quien destacó la falta de correlación entre los aspectos clínicos y patológicos en varios tumores y el hecho de que un número muy elevado de tumores (hasta un 30%) no podía clasificarse siguiendo los criterios de los autores. Paralelamente, autores como Cushing se centraron en clasificaciones clínicas que describían tumores con un pronóstico favorable, por ejemplo, el astrocitoma cerebeloso, que destacaron como diferente del astrocitoma cerebral, a pesar de la similitud histogenética entre ambos. Utilizando datos de estudios del cerebro infantil, los autores describieron tumores quísticos no recurrentes que estaban bien definidos en términos de estadio de crecimiento y cuya clasificación fue muy relevante en su momento. Esta distinción no se basaba meramente en principios histogenéticos y citológicos, sino en datos clínicos, histopatológicos y clínicos. También fueron importantes las contribuciones de Penfield (1931) [6].
Las publicaciones y conferencias de Río Hortega durante la década de 1930 desempeñaron un papel fundamental en la promoción de la clasificación histogenética, en gran parte gracias a técnicas de tinción con plata muy precisas. La mayoría de los autores de este período y posteriores consideraron que su clasificación contenía la colección de imágenes más exhaustiva hasta entonces. La clasificación de Río Hortega no se basaba en hallazgos clínicos o en la localización anatómica, sino en datos morfológicos e histogenéticos, un retorno a la pureza celular como principio organizador fundamental [6].
Tumores del Sistema Nervioso Central: La Contribución de Río Hortega
Durante la etapa inicial de su formación, Río Hortega analizó los tumores cerebrales en cuatro estudios. Uno de ellos fue su tesis doctoral (“Causas y Anatomía Patológica de los Tumores de Encéfalo”), que defendió bajo la dirección de su tutor, Leopoldo López García, entre los años 1911 y 1912 [6].
Río Hortega escribió artículos sobre la histopatología de los carcinomas y del sistema nervioso en pacientes con tumores cerebrales (1911a), la fisiopatología de los tumores cerebrales (1911b), y las anomalías del tejido nervioso y los síntomas generales de los tumores cerebrales [6].
Durante la siguiente fase de su formación, Río Hortega realizó un estudio sobre el glioma subcutáneo de células gigantes, cuyos resultados se publicaron en 1926 [6]. La tercera fase (1928–1936) vio la aparición de sus contribuciones más importantes al campo de la neurooncología. En 1930, publicó una serie de monografías en las que analizaba las características citológicas e histogenéticas de grupos tumorales específicos, comenzando con una consideración detallada de los exoteliomas meníngeos, que discutió e incluyó en el diagnóstico diferencial de lo que entonces se conocía como meningioma de Cushing. Describió variaciones del meningioma, como los tumores xantomatosos y los tumores fasciculares, que habían sido reportados esporádicamente por otros autores. El examen de estas formas histopatológicas le llevó a proponer tres patrones principales: (a) un patrón predominantemente sincitial; (b) un patrón basado en la diferenciación fibrilar del citoplasma que tendía a disponerse en placas y haces; y (c) un patrón que implicaba formas morfológicas más epitelioides y lobuladas. Del mismo modo, describió la formación de acérvulos y cuerpos de psammoma en el meningioma, la glándula pineal y el plexo coloide.
El año 1932 vio la publicación del importante estudio “La estructura y sistematización de los gliomas y paragliomas”, que, con más de 260 páginas y 200 imágenes, fue el fruto de las técnicas que Río Hortega había estado desarrollando utilizando principalmente la tinción con carbonato de plata.
Realizó el estudio utilizando su profundo conocimiento de la histología de la glía y el cerebro y tuvo que buscar la ayuda de neurocirujanos y otros patólogos para recopilar una serie suficientemente grande de muestras de tumores cerebrales para su estudio y clasificación. El neurocirujano francés Clovis Vincent fue de inestimable ayuda durante este período.
Los resultados, que se basaron en datos neuroembriológicos, señalaron cuatro posibles vías evolutivas del epitelio medular primitivo (neuroblastos, glioblastos, pineoblastos y coroideoblastos). Dada la naturaleza considerablemente heterogénea de los tumores cerebrales, Río Hortega pensó que era importante clasificarlos en tipos histológicos con hallazgos embriológicos comunes. Por lo tanto, intentó definir dos grandes grupos de tumores, con énfasis en el linaje histológico y embriológico. El primer grupo comprendía los gliomas y el segundo los paragliomas, que incluían todos aquellos tumores formados por elementos inmaduros o maduros del sistema nervioso y tumores que surgían de los pliegues coroideos y la glándula pineal.
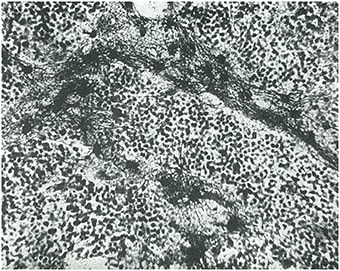
Su tipificación sistemática de los gliomas según el grado de madurez de los componentes celulares o el grado de diferenciación le permitió definir las siguientes entidades (Figura 1):
-
Glioblastoma embrionario o espongioblastoma.
-
Glioblastoma heteromórfico.
-
Glioblastoma isomórfico (Figuras 2, 3).
-
Astroblastoma.
-
Astrocitoma (Figura 4).
-
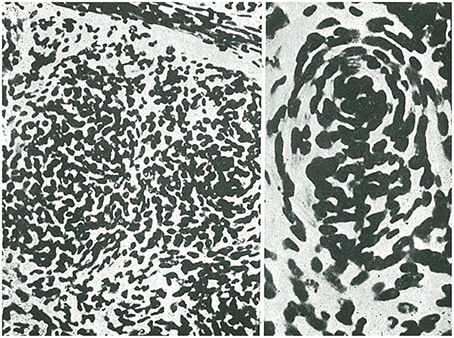
Oligodendroglioma, con una distinción entre oligodendrocitomas y oligodendroblastomas (Figura 5).
-
Glioepiteliomas, que incluían tumores ependimarios (ependimocitoma y ependimoblastoma).
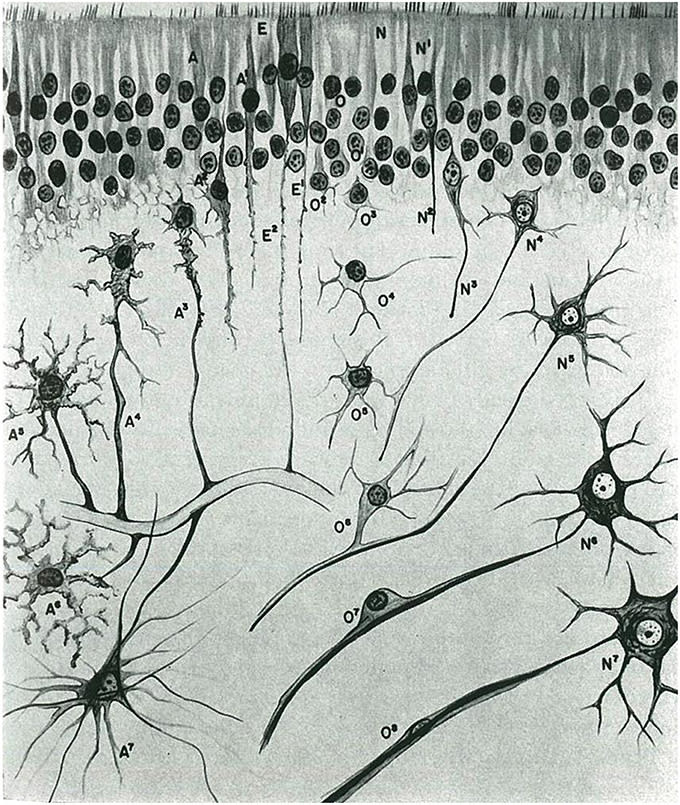
La clasificación de los paragliomas incluía el neuroma de neuroblastos (neuroblastoma), el neurocitoma, los tumores pineales (pinealocitoma, pineoblastoma) y los tumores del plexo coroideo, que denominó corioepiteliomas (Figura 6).

En el Congreso Internacional del Cáncer sobre la Lucha Científica y Social contra el Cáncer, celebrado en Madrid en 1933, ofreció un resumen más extenso de su clasificación en una conferencia basada en 287 páginas de texto (315 incluyendo la bibliografía) y 248 imágenes.
La clasificación, que en cierta medida complementaba las propuestas por Roussy y Oberling y especialmente la propuesta por Bailey y Cushing, difería en áreas importantes, algunas de las cuales merecen ser mencionadas. La clasificación de Río Hortega se basaba en las características citológicas y embriológicas de las células tumorales, independientemente de su localización, y por lo tanto incluía tumores como el meduloblastoma cerebeloso junto con tumores de linaje blástico del interior del cerebro. Esta distinción entre meduloblastomas y otros tumores neuroblásticos o primitivos fue controvertida en su momento y sigue siéndolo hoy en día.
En su conferencia, Río Hortega subrayó la necesidad de una armonización internacional de la nomenclatura aplicada a los tumores del sistema nervioso, como también propusieron Roussy y Oberling. Los grupos que sugirió en la conferencia fueron los siguientes:
-
Tumores que surgen de los pliegues coroideos, la válvula pineal y evaginaciones homólogas del diencéfalo que se desarrollan en el embrión.
-
Tumores que surgen en el parénquima del cerebro y la médula espinal y el sistema visual, que es una prolongación del cerebro.
-
Tumores que surgen en el sistema nervioso simpático, pero no todos los que se desarrollan a partir de simpatogonias.
-
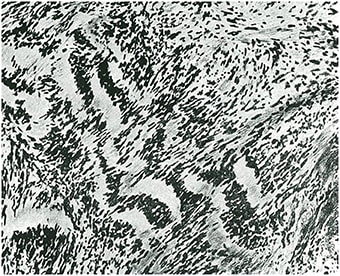
Tumores que surgen en las raíces nerviosas y en los nervios periféricos a partir de células intersticiales o células parenquimatosas, dependiendo de la interpretación de los elementos neoplásicos (Figura 7).
-
Tumores que surgen en las meninges debido a la proliferación de células o a nuevas formaciones vasculares (Figura 8).
-
Tumores que surgen de la hiperplasia en el parénquima de la glándula pituitaria y de células germinales epidérmicas desplazadas e invaginaciones de la bolsa de Rathke.
La cuarta etapa de la vida de Río Hortega (1936-1945), que pasó en el exilio, vio la publicación de varios trabajos sobre el sistema nervioso.
En 1940, tras su estancia en Oxford, realizó un estudio sobre los tumores del nervio óptico. Durante el mismo año, una vez instalado en Argentina, publicó un estudio sobre los neuroblastomas, en el que concluía que no existían tumores del sistema nervioso con células bipotenciales capaces de progresar a neuroblastos o glioblastos y que la mayoría de los llamados meduloblastomas debían denominarse neuroblastomas, que es el término que corresponde a su linaje. Su enfoque fue considerado científico en términos de su interpretación embriológica.
Esta interpretación sigue siendo controvertida hoy en día. El enfoque embriológico no tuvo en cuenta las características histopatológicas. Además, el grupo de los neuroblastomas incluía tumores que se desarrollaban a partir de otros precursores, a saber, los meduloblastos, que se desarrollan en la capa molecular del cerebelo.
Su artículo titulado “Del glioepitelioma al glioblastoma isomorfo”, publicado en 1941, discutía y criticaba el uso del término ependimoma —sugerido por Bailey y Cushing— para los tumores asociados a la pared ependimaria.
En 1943, Río Hortega realizó un estudio citológico de los neurofibromas (también conocidos como lemocitomas), en el que describió las características histológicas de los tumores y los elementos que los caracterizan. Realizó un examen en profundidad de la constitución de estos tumores, discutió la identificación de los principales elementos de las células de Schwann y el origen embrionario de las mismas, y examinó la diferenciación específica de los neurofibromas múltiples y los neurinomas (schwannomas solitarios). Sus hallazgos siguen vigentes en la actualidad.
Añadió nueva información patológica después de trabajos anteriores.
En 1944, Río Hortega informó de los resultados de un extenso estudio sobre los oligodendrogliomas, que clasificó como una variedad ectodérmica gliomatosa caracterizada por células pequeñas con un núcleo esférico. Su descripción del núcleo como “muy redondo” sigue siendo de utilidad hoy en día en el diagnóstico de los oligodendrogliomas. Del mismo modo, observó que los oligodendrocitos tienden a disponerse en patrones densos o difusos y nunca en patrones perivasculares. Río Hortega estableció tres tipos citológicos de oligodendroglioma:
-
(a) aquellos cuyas células tienen un núcleo esférico rodeado por un halo claro característico y envuelto en una pequeña capa de protoplasma que proyecta un número variable de apéndices finos y largos;
-
(b) un tipo más infrecuente de oligodendroglioma, que está formado por grandes oligodendrocitos neoplásicos; y
-
(c) un tipo que incluye tumores con una estructura no uniforme. Como señaló Río Hortega, el oligodendrocito neoplásico evoluciona morfológicamente hasta el punto de que adquiere las características de un astrocitoma.
El año 1944 también es notable por el estudio citológico de Río Hortega sobre los tumores del quiasma y el nervio óptico. Los tumores descritos a este nivel que pueden clasificarse como gliomas, que eran similares a los tumores cerebrales, con un carácter moderadamente expansivo o infiltrativo. Los diversos tipos de células que pueden identificarse para los tumores del nervio óptico y el quiasma incluyen los siguientes:
-
(1) células con núcleos pequeños y redondos;
-
(2) células con núcleos bipolares, fusiformes y largos;
-
(3) células con un citoplasma tripolar y prolongaciones gruesas;
-
(4) células con citoplasma multipolar y prolongaciones fibroides y ondulantes;
-
(5) células con citoplasma multipolar que invaden la vasculatura.
Río Hortega llegó a la conclusión, aunque indefinida, de que existen dos tipos neoplásicos básicos en las formaciones que estudió: uno caracterizado por elementos largos (oligodendrocitos de Schwann) y otro definido por elementos multipolares que le dan la apariencia de astrocitos.
Clasificación de los Tumores del Sistema Nervioso Central Después de Río Hortega
Las clasificaciones actuales de los tumores del sistema nervioso son mixtas, basadas en criterios citológicos e histogenéticos, así como en variantes histopatológicas que tienen importancia clínica y pronóstica.
Los principales estudios publicados después de Río Hortega incluyen el de Kernohan y Sayre (1952), y Miller et al. (1952), quienes comenzaron a graduar los gliomas estableciendo una correlación entre los hallazgos microscópicos, el grado de malignidad y el pronóstico.
En 1965, Zülch destacó la importancia de otros factores, como la supervivencia del paciente, e incluyó el concepto de malignidad clínica. Finalmente, la primera clasificación de la Organización Mundial de la Salud se publicó en 1979 y clasificó los tumores de la siguiente manera:
-
Tumores de tejido neuroepitelial, incluyendo astrocitoma, glioblastoma multiforme, oligodendroglioma, ependimoma, pinealocitoma, meduloblastoma, gangliocitoma, ganglioglioma y neuroblastoma.
-
Tumores meníngeos, como meningioma y sarcoma meníngeo.
-
Tumores de las células de las vainas nerviosas, como neurinoma y neurofibroma.
-
Linfoma cerebral primario.
-
Tumores que surgen en los vasos sanguíneos, como el hemangioblastoma.
-
Tumores de células germinales, como germinoma y teratoma.
-
Tumores metastásicos.
-
Tumores malformativos y lesiones similares a tumores, como craneofaringioma, quiste epidermoide, quiste dermoide y quiste coloide del tercer ventrículo.
-
Extensiones locales de tumores regionales, como el tumor del glomus yugular y el cordoma.
-
Tumores de la hipófisis anterior, como el adenoma hipofisario.
-
Tumores no clasificados.
Esta clasificación sirve de base para las ediciones posteriores de la clasificación de la Organización Mundial de la Salud hasta el año 2007 y los subgrupos que se están incorporando actualmente. Las nuevas adiciones más notables son las siguientes:
-
Variantes de astrocitomas de grado 1, como el astrocitoma fibrilar, protoplásmico y gemistocítico.
-
Astrocitoma pilocítico como entidad independiente.
-
Astrocitoma subependimario de células gigantes.
-
Astroblastoma.
-
Astrocitoma anaplásico (maligno).
También se hace una distinción entre los tumores oligodendrogliales y los tumores oligoastrocíticos .
Dentro de los tumores ependimarios y los tumores del plexo coroideo, es importante distinguir entre las variantes de los ependimomas, como el ependimoma mixopapilar, el ependimoma papilar, el subependimoma y el ependimoma anaplásico. A nivel del plexo coroideo, debemos distinguir entre el papiloma del plexo coroideo y el carcinoma del plexo coroideo.
Los tumores neuronales incluyen variantes como gangliocitoma, ganglioglioma, ganglioneuroblastoma, gangliocitoma, ganglioglioma anaplásico (maligno) y neuroblastoma.
Entre los tumores poco diferenciados y embrionarios es importante identificar el glioblastoma (con sus dos subvariantes, glioblastoma con componente sarcomatoso y glioblastoma de células gigantes), el meduloblastoma, el meduloepitelioma, el espongioblastoma polar primitivo y la gliomatosis cerebral.
La clasificación abarca los tumores de los tejidos meníngeos y afines, como el meningioma, con al menos 11 variantes morfológicas según el predominio del componente de Schwann, angiomatoso y papilar. Del mismo modo, la variante anaplásica (maligna) del meningioma es una entidad distinta.
La clasificación todavía incluye tumores vasculares (p. ej., hemangioblastoma y una variante maligna conocida como carcinoma monocelular), linfoma maligno primario y varias variantes de tumores de células germinales. El grupo previamente citado de tumores malformativos y lesiones similares a tumores se amplía para incluir quistes enterógenos, lipoma, hamartoma neuronal hipotalámico, heterotopía glial nasal (glioma nasal), así como diversas malformaciones vasculares (telangiectasia capilar, malformaciones arteriovenosas y enfermedad de Sturge-Weber).
La clasificación de 2007 sigue incluyendo nuevas entidades, principalmente condiciones anatómico-clínicas en las que es muy importante distinguir entre gliomas con un alto y bajo grado de malignidad según criterios citológicos. Los nuevos tipos de glioma de bajo grado descritos incluyen los gliomas angiocéntricos, que son variantes de los tumores glioneuronales (p. ej., tumores formadores de rosetas o papilares), y las variantes citológicas de los tumores de la hipófisis anterior (p. ej., pituicitoma y oncocitoma de células fusiformes). También podemos distinguir entre los tumores pilocíticos y sus variantes pilomixoides, que tienen un peor pronóstico clínico.
En los próximos años, será necesario añadir las anomalías moleculares subyacentes a la transformación y malignidad de estos tumores. Se espera que nuestro conocimiento aumente gracias a la amplificación de genes como el EGFR en el glioblastoma, la pérdida de alelos en los cromosomas 1p y 19p en el oligodendroglioma y las mutaciones en genes como p53 e IDH1 en el astrocitoma de bajo grado que progresa a astrocitoma maligno. La heterogeneidad intra e intertumoral podría entenderse como resultado de las células madre cancerosas y la acumulación de diversas anomalías moleculares.
Como ha ocurrido con otros tipos de tumores, especialmente el linfoma, cuyas clasificaciones se han basado durante décadas meramente en criterios morfológicos o clínicos, un enfoque conjunto de la clasificación es probablemente el más adecuado para la práctica clínica. Las anomalías citológicas, la localización y las características histopatológicas podrían facilitar un estudio más profundo de los diversos tipos de tumores. Es importante recordar que el objetivo principal de cualquier clasificación es que la información que proporciona sea de utilidad en la práctica clínica. Solo así el paciente podrá recibir el mejor y más personalizado tratamiento posible.
Financiación
Fondo de Investigaciones Sanitarias (11/00185), Redes Temáticas de Investigación Cooperativa en Salud (Ref. RD06/0020/1020).
Glosario Terminológico de Neuro-Oncopatología
Para garantizar la máxima precisión y claridad, se presenta a continuación una tabla con los términos técnicos clave utilizados en este artículo, su traducción al español y la justificación basada en fuentes médicas y científicas autorizadas.
Término en InglésTraducción al EspañolJustificación y NotasCentral Nervous System (CNS)Sistema Nervioso Central (SNC)Término estándar utilizado en publicaciones médicas y bases de datos como MedlinePlus y el Instituto Nacional del Cáncer (NCI) [12, 13].NeuropathologyNeuropatologíaDisciplina que estudia las enfermedades del sistema nervioso. Término oficial según el Diccionario de Términos Médicos (DTME) de la Real Academia Nacional de Medicina de España y los Descriptores en Ciencias de la Salud (DeCS) [14, 15].Histology / CytologyHistología / CitologíaDisciplinas fundamentales. Histología: estudio de los tejidos. Citología: estudio de las células. La distinción es clave para el enfoque de Río Hortega [16, 17].Silver Staining MethodMétodo de tinción con plataTraducción general [18]. El método específico de Río Hortega es el “método del carbonato de plata” [10, 11].Cell Lineage / HistogenesisLinaje celular / HistogénesisConceptos centrales de la clasificación de Río Hortega, basados en el origen y desarrollo embrionario de las células tumorales [19, 20].GliomaGliomaTérmino general para tumores que surgen de células gliales. Se mantiene el término en español, ya que es de uso internacional [21, 22, 23].AstrocytomaAstrocitomaTipo de glioma que surge de los astrocitos. Se mantiene el término [24, 25].GlioblastomaGlioblastomaAstrocitoma de alto grado (Grado IV). Término estándar internacional [25, 26, 27].Oligodendroglia / MicrogliaOligodendroglía / MicroglíaTipos de células gliales descubiertas/caracterizadas por Río Hortega. Los términos son idénticos en español [8, 9].MedulloblastomaMeduloblastomaTumor embrionario común en el cerebelo, especialmente en niños. Término estándar [28, 29].EpendymomaEpendimomaTumor que surge de las células ependimarias que recubren los ventrículos. Término estándar [21].Choroid Plexus PapillomaPapiloma del plexo coroideoTumor benigno que surge del plexo coroideo. Traducción literal y estándar [30, 31].CraniopharyngiomaCraneofaringiomaTumor benigno cerca de la hipófisis (glándula pituitaria). Término estándar [32, 33].
Referencias
-
Bailey, P. (1924). A study of tumors arising from ependymal cells. Arch. Neurol. Psychiatry 11, 1. doi: 10.1001/archneurpsyc.1924.02190310007001
-
Bailey, P., and Bucy, P. (1929). Oligodendrogliomas of the brain. J. Pathol. Bacteriol. 32, 735.
-
Bailey, P., and Cushing, H. (1925). Medulloblastoma cerebelli. A commontype of mid cerebellar glioma of childhood. Arch. Neurol. Psychiatry 14, 192.
-
Bailey, P., and Cushing, H. A. (1926). A Classification of the Tumours of the Glioma Group on a Histogenetic Basis, with a Correlated Study of Prognosis. Philadelphia, PA; London; Montreal, QC: J. B. Lippincott Company.
-
Cornil, L. (1924). Considerations anatomiques sur les nerfs crâniens. Rev. Med. 52, 682.
-
Cushing, H. (1917). Tumors of the Nervus Acusticus and the Syndrome of the Cerebellopontile Angle. Philadelphia, PA; London: W. B. Saunders company.
-
Diaz, P. C. (1985). Una contribucion a la Ciencia Histologica: La obra de Do Pio del Rio-Hortega. Madrid: CSIC.
-
Globus, J. H., and Strauss, I. (1925). Spongioblastoma multiformea primary malignant form of brain neoplasm: its clinical and anatomic features. Arch. Neurol. Psychiatry 14, 139–191.
-
Kernohan, J. W., and Ody, F. A. (1932). Classification histologique des gliomes de la moelle épiciére et du filum terminale. Schweiz. Arch. Neurol. Psychiatry 29, 113.
-
Kernohan, J. W., Woltmann, H. W., and Adson, A. W. (1931). Intramedullary tumors of the spinal cord. A review. Arch. Neurol. Psychiatry 25, 679.
-
Kernohan, J. W., and Sayre, G. P. (1952). “Tumors of the central nervous system,“ in Atlas of Tumor Pathology, fasc. 35, Washington, DC: Armed Forces Institute of Pathology.
-
Lhermitte, J., and Dumas, R. (1916). La ganglioneuromatose disseminée, type anatomique du syndrome de Recklinghausen. Rev. Neurol. 29, 579.
-
Llombart Rodríguez, A. (1965). La personalidad humana y la significación histoneuropatológica de don Pío del Río-Hortega. Rev. Oncología II, 29–36.
-
Louis, D. N., Ohgaki, H., Wiestler, O. D., Cavenee, W. K., Burger, P. C., Jouvet, A., et al. (2007). The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109. doi: 10.1007/s00401-007-0243-4
-
Miller, R. H., Craig, W. M., and Kernohan, J. W. (1952). Supratentorial tumors among children. AMA Arch. Neurol. Psychiatry 68, 797–814.
-
Obrador, S. (1965). Pío del río-hortega. Rev. Esp. Oncología II, 12–13.
-
Ortiz de Picon, J. M. (1983). Pio del Rio-Hortega. Un estudio blibliográfico. ARch. Neurobiol. 46, 209–226.
-
Penfield, W. (1931). A paper on classification of brain tumors and its pratical application. Br. J. Med. 3660, 337.
-
Pérez-Cerdá, F., Sánchez-Gómez, M. V., and Matute, C. (2015). Pío del Río Hortega and the discovery of the oligodendrocytes. Front. Neuroanat. 9:92. doi: 10.3389/fnana.2015.00092
-
Pineda, A., Russell, G. V., and Kenneth, M. (1962). The Microscopy Anatomy of tumors of the Central and Peripheral Nervous system. Springfiled, IL: CC Thomas Publisher.
-
Polak, M. (1947). Pío del Río-Hortega (1882-1945). Arch. Hist. Norm. 3, 337–415.
-
Ribbert, H. (1910). Neuroepithel in Gliomen. Zentralbl. Allg. Path. 21, 145.
-
Ribbert, H. (1918). Über das spongioblastom und das Gliom. Virchow Arch. 225, 195.
-
Río-Hortega, P. (1911a). Fisiopatología de los Tumores del Encéfalo. Valladolid: La Clínica Castellana.
-
Río-Hortega, P. (1911b). Histopatología Nerviosa en los Tumores del Encéfalo. Valladolid: La Clínica Castellana.
-
Río-Hortega, P. (1912). Alteraciones del Tejido Nervioso y Síntomas Generales de los Tumores del Encéfalo. Valladolid: La Clínica Castellana.
-
Río-Hortega, P. (1914c). Conexiones Entre el Tejido Conjuntivo y Las Células del Carcinoma. Bol. Soc. Esp. Biol. III, 123–124.
-
Río-Hortega, P. (1914a). Contribución al Conocimiento de la Fina Textura de las Células Cancerosas. Las Epiteliofibrillas. Madrid: Trab. Del Lab. De Inv. Biol. De la Univ.
-
Río-Hortega, P. (1914b). Sobre la existencia de epiteliofibrillas en las células cancerosas. Bol. Soc. Esp. Biol. III, 124–128.
-
Río-Hortega, P. (1926). Glioma subcutáneo de células gigantes. Bol. Soc. Esp. Biol., XII, 1–9.
-
Río-Hortega, P. (1930c). Localización de las Concreciones Calcáreas en los Endoteliomas Meníngeos y Gliomas.
-
Río-Hortega, P. (1930a). Para el mejor conocimiento histológico de los meningoexoteliomas. Arch. Esp. De Oncología, I., 477–570.
-
Río-Hortega, P. (1930b). Sobre la Formación de los Acérvuli en Plexos Coroideos, Glándula Pineal y Psamomas. Libro homenaje a Goyanes, febrero.
-
Río-Hortega, P. (1932). Estructura y Sistematización de los Gliomas y Paragliomas. Arch. Esp. De Oncología, I. I., 411–677.
-
Río-Hortega, P. (1933a). Anatomía microscópica de los tumores del sistema nervioso central y periférico. Trabajos del Lab. De Histopat. De la Junta para Ampliación de Estudios, número 103.
-
Río-Hortega, P. (1933b). Arte y artificio de la ciencia histológica. en Residencia Revista de la Residencia de Estudiantes IV, 198–200.
-
Río-Hortega, P. (1940a). Neuroblastomas. Buenos Aires: Bol. Acad. Nal. Med.
-
Río-Hortega, P. (1940b). Discussion on tumors of the optic nerve. Proc. R. Med. XXXIII, 686.
-
Río-Hortega, P. (1940c). Neuroblastomas. Buenos Aires: Bol. Acad. Nal. Med.
-
Río-Hortega, P. (1941a). Del glioepitelioma al glioblastoma isomorfo. Arch. Soc. Arg. Anat. Norm. Y Pat. III, 473.
-
Río-Hortega, P. (1941b). Nomenclatura y clasificación de los tumores del sistema nervioso. Arch. Arg. Neurol. XXIV, 7.
-
Río-Hortega, P. (1942). Caracteres e interpretación de las células específicas de los Neurinomas (Schwannomas). Arch. Soc. Arg. Anat. Norm. Pat. IV, 103.
-
Río-Hortega, P. (1943). Estudio citológico de los neurofibromas de Recklinghausen/lemnocitomas).- Células específicas. Arch. Hist. Norm. Pat. I, 373–414.
-
Río-Hortega, P. (1944a). Contribución al conocimiento citológico de los oligodendrogliomas. Arch. Hist. Norm. Pat. II, 267–305.
-
Río-Hortega, P. (1944b). Contribución al conocimiento citológico de los tumores del nervio y quiasma ópticos. Arch. Hist. Norm. Pat. II, 307–358.
-
Río-Hortega, P., and y Costero, I. (1928). Ramificación endocapsular neuronoide de las células condromatosas. Bol. Soc. Esp. Bio. XIII, 97–105.
-
Río-Hortega, P., and y Jiménez de Asúa, F. (1921). Sobre la fagocitosis en los tumores y en otros procesos patológicos. Arch. Card. Y Hemat. II, 161–220.
-
Río-Hortega, P., Prado, J., and y Polak, M. (1943). Sinticio y diferenciaciones citoplasmáticas de los meningoexoteliomas. Arch. Hist. Norm. Pat. II, 125–170.
-
Río-Hortega, P., and y Álvarez Cascos. (1930). Variaciones histológicas del cáncer de la piel. Arch. Esp. De Oncología.
-
Roussy, G., and Oberling, C. (1932). Histologic classification of tumors of the central nervous system. Arch. Neurol. Psychiatry 27, 1281.
-
Scherer, H. J. (1933). Zur Frage der Zusammenhanges zwischen Neurofibromatose (Recklinghausen) und umschriebenen Riesenwuchs. Virchow’s Arch. 289, 127.
-
Simon, J. (1874). Re’ forme de L’enseignement Secondaire. Paris; Hachette: Simon J.
-
Tremblay, M. È., Lecours, C., Samson, L., Sánchez-Zafra, V., and Sierra, A. (2015). From the Cajal alumni Achúcarro and Río-Hortega to the rediscovery of never-resting microglia. Front Neuroanat. 9:45. doi: 10.3389/fnana.2015.00045
-
Zülch, K. J. (1979). Histological Typing of Tumours of the Central Nervous System. Geneva: World Health Organization.
-
Zülch, K. (1965). Some characteristics of brain tumors related to the age, site and sex of the tumor patients. Zentralbl. Chir. 90, 890–898.


Comentarios
Para activar los comentarios: ve a giscus.app, introduce el repositorio
joseadserias-dotcom/cajal-digitaly reemplaza los IDs ensrc/layouts/Articulo.astro.